Metal-free catalysis and biocatalysis
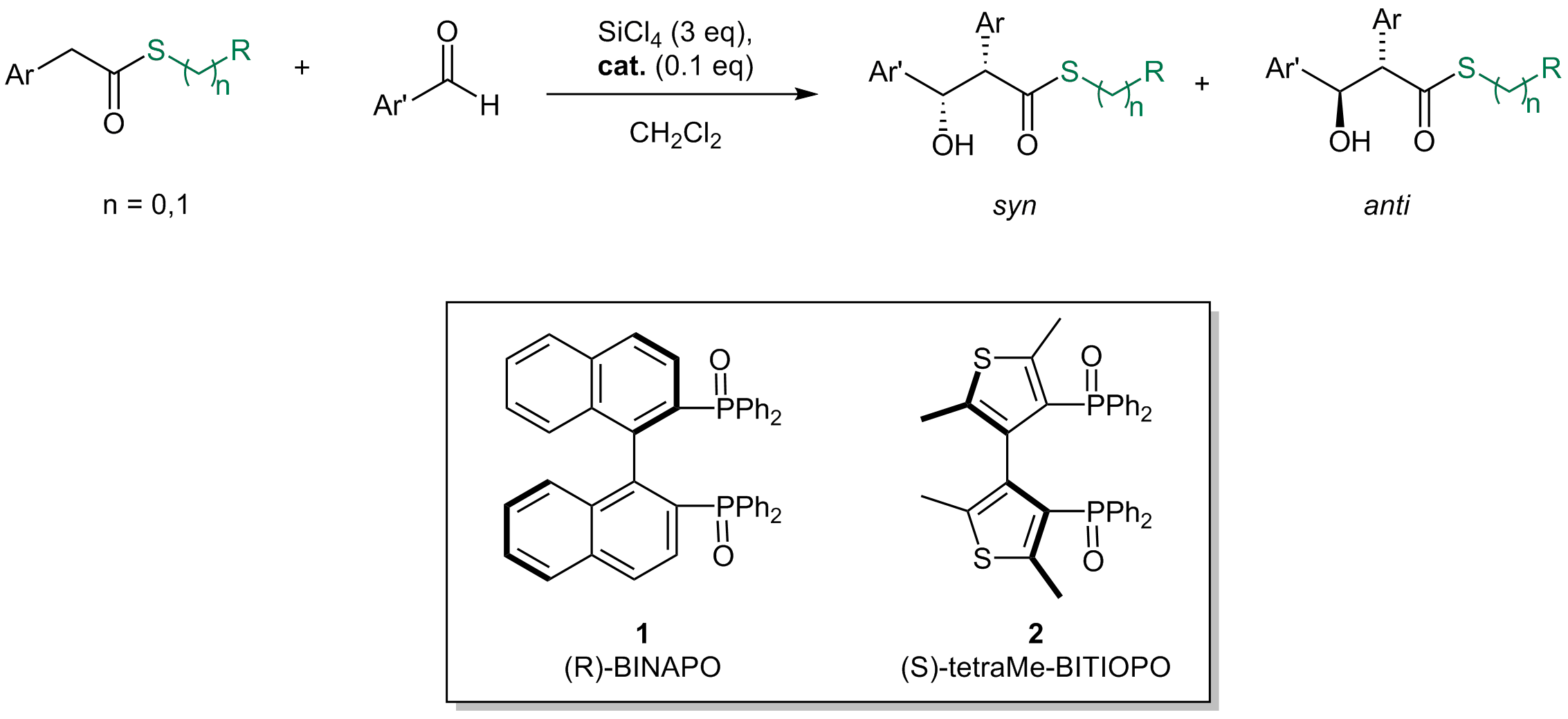
A one pot protocol to convert nitro-arenes into N-aryl amides
E. Massolo, M. Pirola, A. Puglisi, L. S. Rossi, M. Benaglia RSC Advances, 2020, 10, 4040-4044
[Link]
Abstract:
A two-step one pot, experimentally simple protocol, based on readily available and inexpensive reagents allowed to convert
nitro-arenes directly to N-aryl amides. A metal-free reduction of the nitro group, mediated by trichlorosilane, followed
by the addition of an anhydride afforded the corresponding N-aryl carboxyamide, that was isolated after a simple aqueous work up
in good-excellent yields. When the methodology was applied to the reaction with γ-butyrolactone, the desired N-aryl butanamide
derivative was obtained, featuring a chlorine atom at the γ-position, a functionalized handle that can be used for further
synthetic manipulation of the reaction product. Such intermediate has been already employed as key advanced precursor of
pharmaceutically active compounds.
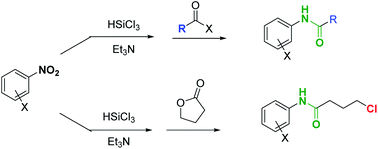
Enantioselective synthesis of 3,4-dihydropyran-2-ones via PTC addition-cyclisation of acetylacetone to cinnamic thioesters
D. Destro, C. Bottinelli, L. Ferrari, D.C.M. Albanese, G. Bencivenni, M.W. Gillick-Healy, B.G. Kelly, M.F.A. Adamo J. Org. Chem., 2020
[Link]
Abstract:
Herein, we present the first example of synthesis of 3,4-dihydropyran-2-ones from cinnamic thioesters via a stereoselective
phase-transfer-catalyzed domino Michael–cyclization reaction with acetylacetone. The reaction proceeded under the catalysis
of Cinchona-derived quaternary ammonium phenoxide that, in combination with inorganic bases, provided 3,4-dihydropyran-2-ones
in yields of up to 93% and enantioselectivities of up to 88% enantiomeric excess.
Synthesis of Ribavirin, Tecadenoson, and Cladribine by Enzymatic Transglycosylation
M. Rabuffetti, T.Bavaro, R. Semproli, G. Cattaneo, M. Massone, C.F. Morelli, G. Speranza, D. Ubiali
Catalysts, 2019, 9 335 [Link]
Abstract:
Despite the impressive progress in nucleoside chemistry to date, the synthesis of nucleoside analogues is still a challenge.
Chemoenzymatic synthesis has been proven to overcome most of the constraints of conventional nucleoside chemistry.
A purine nucleoside phosphorylase from Aeromonas hydrophila (AhPNP) has been used herein to catalyze the synthesis
of Ribavirin, Tecadenoson, and Cladribine, by a “one-pot, one-enzyme” transglycosylation, which is the transfer of the
carbohydrate moiety from a nucleoside donor to a heterocyclic base. As the sugar donor, 7-methylguanosine iodide and its
20-deoxy counterpart were synthesized and incubated either with the “purine-like” base or the modified purine of the three
selected APIs. Good conversions (49–67%) were achieved in all cases under screening conditions. Following this synthetic
scheme, 7-methylguanine arabinoside iodide was also prepared with the purpose to synthesize the antiviral Vidarabine by a
novel approach. However, in this case, neither the phosphorolysis of the sugar donor, nor the transglycosylation reaction
were observed. This study was enlarged to two other ribonucleosides structurally related to Ribavirin and Tecadenoson, namely,
Acadesine, or AICAR, and 2-chloro-N6-cyclopentyladenosine, or CCPA. Only the formation of CCPA was observed (52%). This
study paves the way for the development of a new synthesis of the target APIs at a preparative scale. Furthermore, the
screening herein reported contributes to the collection of new data about the specific substrate requirements of AhPNP.
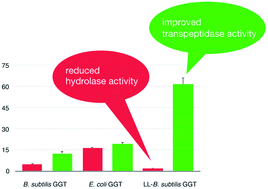
Effect of the Inserted Active-site-covering Lid Loop on the Catalytic Activity of a Mutant B. Subtilis γ-Glutamyltransferase (GGT)
M. Massone, C. Calvio, M. Rabuffetti, G. Speranza, C.F. Morelli
RSC Adv., 2019, 9 34699 [Link]
Abstract:
γ-Glutamylpeptides are compounds derived from the acylation of an amino acid or a short peptide by the γ-carboxyl carbon
of the side chain of glutamic acid. Due to their altered chemico-physical and organoleptic properties, they may be
interesting substitutes or precursors of parent compounds used in pharmaceutical, dietetic and cosmetic formulations.
Some of them are naturally occurring flavor enhancers or are endowed with biological activities. Enzymatic approaches to the
synthesis of γ-glutamyl derivatives based on the use of γ-glutamyltransferases (GGTs, EC 2.3.2.2) have been proposed, which
should be able to alleviate the problems connected with the troublesome and low-yielding extraction from natural sources or
the non-economical chemical synthesis, which requires protection/ deprotection steps. With the aim of overcoming the current
limitations in the use of GGTs as biocatalysts, a mutant GGT was investigated. The mutant GGT was obtained by inserting the
active-sitecovering lid loop of the E. coli GGT onto the structure of B. subtilis GGT. With respect to the wild-type enzyme,
the mutant showed a more demanding substrate specificity and a low hydrolase activity. These results represent an attempt to
correlate the structural features of a GGT to its different activities. However, the ability of the mutant enzyme to catalyze
the subsequent addition of several γ-glutamyl units, inherited by the parent B. subtilis GGT, still represents a limitation
to its full application as a biocatalyst for preparative purposes.
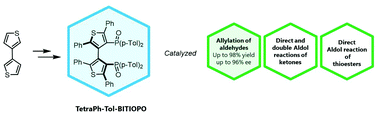
TetraPh-Tol-BITIOPO: a new atropisomeric 3,3’-bithiophene based phosphine oxide as an organocatalyst in Lewis base-catalyzed Lewis acid mediated reactions
V. M. Abbinante, M. Benaglia, S. Rossi; T. Benincori, R. Cirili, M. Pierini Org Biomol Chem, 2019, 17, 7474-7481.[Link]
Abstract:
A new chiral phosphine oxide based on a 3,3’-bithiophene scaffold (TetraPh-Tol-BITIOPO) was synthesized, fully characterized and
separated into antipodes through chiral HPLC. This new compound was successfully employed as an organocatalyst in Lewis base-catalyzed
Lewis acid mediated reactions involving trichlorosilyl compounds. The new atropisomeric catalyst was able to promote the allylation
of aldehydes with allyltrichlorosilane in up to 98% yield and up to 96% enantiomeric excess (ee), and the direct aldol reaction to
afford β-hydroxy ketones and β-hydroxy thioesters, with good chemical yields and modest stereochemical efficiency. Computational
studies helped to elucidate and to rationalize the stereochemical outcome of the reactions catalyzed by TetraPh-Tol-BITIOPO that was
found to favour the formation of the isomer with an opposite absolute configuration in comparison with the products obtained with the
previously reported 3,3’-bithiophene-based catalyst.
Evidences on the Role of the Lid Loop of γ-glutamyltransferases (GGT) in Substrate Selection,
C. Calvio, F. Romagnuolo, F. Vulcano, G. Speranza, C.F. Morelli
Enz. Microb. Technol. , 2018, 114 55-62 [Link]
Abstract:
γ-Glutamyltransferase (GGT) catalyzes the transfer of the γ-glutamyl moiety from a donor substrate such as glutathione to water
(hydrolysis) or to an acceptor amino acid (transpeptidation) through the formation of a γ-glutamyl enzyme intermediate. The vast
majority of the known GGTs has a short sequence covering the glutamate binding site, called lidloop. Although being conserved
enzymes, both B. subtilis GGT and the related enzyme CapD from B. anthracis lack the lid loop and, differently from other GGTs
, both accept poly-γ-glutamic acid (γ-PGA) as a substrate. Starting from this observation, in this work the activity of an
engineered mutant enzyme containing the amino acid sequence of the lid loop from E. coli GGT inserted into the backbone of B.
subtilis GGT was compared to that of the lid loop-deficient B. subtilis GGT and the lid loop-carrier E. coli GGT. Results
indicate that the absence of the lid loop seems not to be the sole structural feature responsible for the recognition of a
polymeric substrate by GGTs. Nevertheless, time course of hydrolysis reactions carried out using oligo-γ-glutamyl glutamines
as substrates showed that the lid loop acts as a gating structure, allowing the preferential selection of the small glutamine
with respect to the oligomeric substrates. In this respect, the mutant B. subtilis GGT revealed to be more similar to E. coli
GGT than to its wild-type counterpart. In addition, the transpeptidase activity of the newly produced mutant enzyme revealed
to be higher with respect to that of both E. coli and wild-type B. subtilis GGT. These findings can be helpful in selecting
GGTs intended as biocatalysts for preparative purposes as well as in designing mutant enzymes with improved transpeptidase
activity.
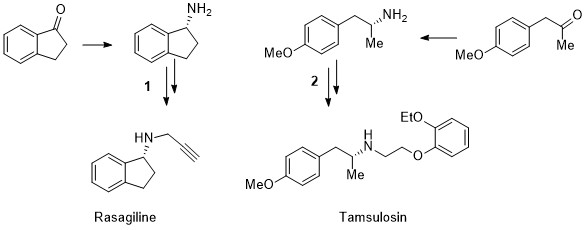
A stereoselective, catalytic strategy for the in-flow synthesis of advanced precursors of Rasagiline and Tamsulosin.
D. Brenna, M. Pirola, L. Raimondi, A. J. Burke, M. Benaglia Bioorg. Med. Chem, 2017, doi:10.1016/j.bmc.2017.01.023 [Link]
Abstract:
The diastereoselective, trichlorosilane-mediate reduction of imines, bearing different and removable chiral auxiliaries, in
combination either with achiral bases or catalytic amounts of chiral Lewis bases, was investigated to afford immediate precursors of
chiral APIs (Active Pharmaceutical Ingredients). The carbon-nitrogen double bond reduction was successfully performed in batch and in
flow mode, in high yields and almost complete stereocontrol. By this metal-free approach, the formal synthesis of Rasagiline and
Tamsulosin was successfully accomplished in micro(meso) flow reactors, under continuous flow conditions. The results represent a new,
important step towards the development of automated processes for the preparation of enantiopure biologically active compounds.
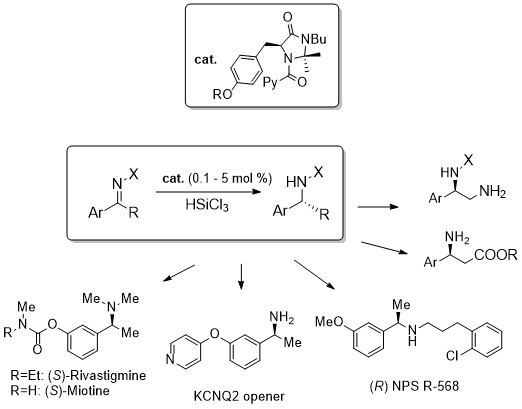
A new class of low-loading catalysts for a highly enantioselective, metal-free imine reduction of wide general applicability
D. Brenna, R. Porta, E. Massolo, L. Raimondi, M. Benaglia ChemCatChem, 2017, 9, 941-945. [Link]
Abstract: A new class of chiral Lewis bases for the enantioselective HSiCl3-mediated reduction of imines has been
developed. Through an extensive catalyst structure optimization, an extremely active species was identified, able to promote the
reduction of a large variety of functionalized substrates in high yields and enantioselectivities typically above 90% with catalyst
loading as low as 0.1-1% mol. The simple experimental procedure, the low cost of the reagents, the mild reaction conditions,
and straightforward isolation of the product, make the methodology attractive also for large scale applications. Its synthetic
potentiality was demonstrated by the preparation of advanced intermediates of important APIs, used in the treatment of Alzheimer
and Parkinson diseases, hyperparathyroidism, neuropathic pains and neurological disorders.The reaction has also been successfully
performed in flow reactors, thus demonstrating the possibility to realize in continuo processes yielding multigram quantities of
product
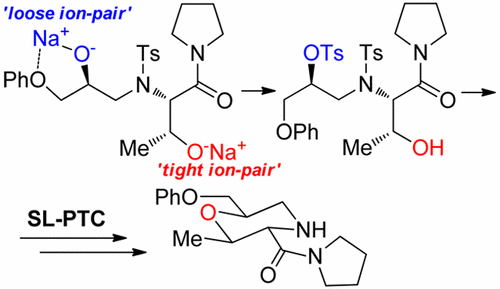
Regioselective O-Sulfonylation of N,N-Bis(2-hydroxyalkyl)tosylamides as a Synthetic Key Step to Enantiopure Morpholines
F. Foschi, D. Albanese, I. Pecnikaj, A. Tagliabue, M. Penso Org. Lett., 2017, 19, 70-73. [Link]
Abstract: The synthesis of enantiopure 2,6-disubstituted morpholines was realized through sequential ring opening of two
different optically pure oxiranes by a tosylamide, under solid–liquid phase-transfer catalysis (SL-PTC) conditions, mono-O-sulfonylation
of the resulting tosylamido-2,2′-diol, and cyclization to the morpholine. The crucial step, the regioselective formation of the
monosulfonate, was controlled by taking advantage of the different stereo, electronic, and coordination properties of the
oxirane-derived side chains in the diol backbone. As an application of this protocol, a new morpholine-3-carboxamide was synthesized
starting from threonine.
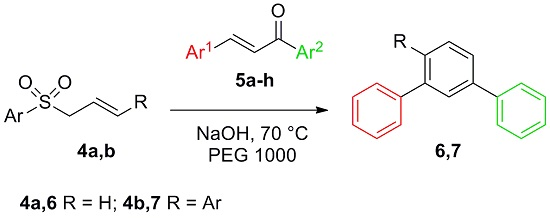
Solventless Synthesis of Quaterphenyls and Terphenyls from Chalcones and Allylsulfones under Phase Transfer Catalysis Conditions
D. C. M. Albanese, S. Brunialti D. Destro Catalysts, 2016, 9, 142. [Link]
Abstract: Easily available chalcones and allyl sulfones along with cheap solid NaOH and polyethyleneglycol (PEG) 1000 have been used
to directly generate the meta-terphenyl or quaterphenyl motifs under Phase Transfer Catalysis solventless conditions. The new approach provides
an economic and environmentally friendly solution to removal of hazardous bases as well as organic solvents.
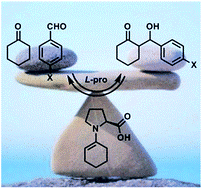
Kinetics versus thermodynamics in the proline catalyzed aldol reaction
M. Orlandi, M. Benaglia, M. Ceotto Chem. Sci., 2016 [Link]
Abstract: In this paper the equilibrium properties of the proline catalyzed aldol reaction was studied. The use
of well-established methodologies, like reaction progress kinetic analysis and linear free energy relationship analysis, led to
the quantification of the reaction reversibility and to its correlation with the substrate electronic activation. Due to these
experimental observations, common computational approaches based on a one way transition state analysis become unsuitable.
Therefore, a computational model based on the integration of a system of kinetic differential equations associated to the multiple
equilibrium reactions was proposed. Such a model was found to successfully rationalize the chemical and stereochemical outcomes
of this paradigmatic reaction for the first time.
Phosphine Oxide Catalyzed, Tetrachlorosilane-Mediated Enantioselective Direct Aldol Reactions of Thioesters
S. Rossi, R. Annunziata, F. Cozzi, L. Raimondi, Synthesis, 2015, [Link]
Abstract:
The stereoselective direct aldol reaction of S-phenyl thioesters and aromatic aldehydes promoted by tetrachlorosilane was realized
for the first time; the proposed mechanism involves the formation of a chiral cationic hypervalent silicon species and requires the
presence of catalytic amounts of a Lewis base, like a chiral phosphine oxide. The reaction affords syn ßhydroxy thioesters as major
isomers in good yields, high diastereoselectivity (up to 99:1), and up to 92% enantiomeric excess. The absolute configuration of the
major isomer was established by converting the product into the corresponding ßhydroxy ester. The scope of the reaction was also
investigated by employing differently substituted thioesters in combination with different aromatic aldehydes.